





Desde hace unos años, el desarrollo de la supercomputación y de la Inteligencia Artificial (AI) ha alcanzado una velocidad imparable, suscitando un enorme interés -así como no poca preocupación e inquietud- entre los expertos, las autoridades y la opinión pública.
Pero junto a sus sombras, la ciencia y la investigación científica, en prácticamente todas las áreas y disciplinas, se están beneficiando de manera espectacular de la supercomputación y la AI, generando avances de gran y beneficioso impacto para la humanidad.
En España, un centro es vanguardia en este campo. El Barcelona Supercomputing Center – Centro Nacional de Supercomputación (BSC-CNS), dependiente de la Universitat Politécnica de Catalunya, alberga las computadoras más potentes de España y algunas de las del mundo. El Mare Nostrum 4 es el ordenador más potente de España y uno de los más potentes del mundo. Es capaz de hacer 14.000 billones de operaciones por segundo. Y su nueva versión, el Mare Nostrum 5, será aún más fugaz, pudiendo ejecutar 200.000 billones de operaciones por segundo.
Entrevistamos a Alfonso Valencia (Sevilla, 1959), pionero de la biocomputación en España. Este investigador de larga trayectoria en la biología molecular dirige el departamento de Ciencias de la Vida del BCS-CNS, también el Instituto Nacional de Bioinformática (INB-ISCIII). Es asimismo miembro fundador de la International Society for Computational Biology (ISCB) y de la Organización Europea de Biología Molecular (EMBO).
.
Ustedes en el Barcelona Supercomputing Center (BSC) montan un caballo muy veloz. El Mare Nostrum 4 es el ordenador más potente de España y uno de los más potentes del mundo. Es capaz de hacer 14.000 billones de operaciones por segundo. Y su nueva versión, el Mare Nostrum 5, será aún más fugaz, pudiendo ejecutar 200.000 billones de operaciones por segundo. Una enorme capacidad de computación que el BSC pone a disposición de múltiples disciplinas, desde su campo, la biomedicina, a la astrofísica. ¿Es así? ¿Qué tipo de investigaciones se llevan a cabo en el BSC?
El Barcelona Supercomputing Center forma parte del Centro Nacional de Supercomputación, es una infraestructura nacional y europea, una de las grandes instalaciones científicas del continente, como son los sincrotrones del CERN o los grandes telescopios de Canarias.
Y como bien dices, el Mare Nostrum era muy potente cuando se instalo, pero los grandes ordenadores, como otras tecnologías, tienen una obsolescencia muy rápida. Y ahora estamos instalando el Mare Nostrum 5 que es 30 veces más potente, gracias a una inversión de unos 220 millones de eurps a medias entre la UE y España. Es un sólo ordenador, pero tiene una memoria compartida con otros. Su hardware son cuatro ordenadores, dos de ellos ya están instalados: uno de ellos es convencional, y otro es de los que se usan para generar Inteligencia Artificial. Cuando esté plenamente operativo, tendrá la capacidad de unas mil CPUs (unidad central de procesamiento, el «motor» de cualquier ordenador), unas de las capacidades de procesamientos más grandes del mundo. Pero no nos preocupa el ránking, sino que sea un ordenador flexible y útil.
Funcionamos a través de proyectos, europeos y nacionales. Cuando un equipo pide poder utilizar los superordenadores, se evalua por un consejo de expertos, y si se autoriza, se usan los Mare Nostrum o sus equivalentes: uno está en Finlandia y otro en Italia. En España también tenemos otros ordenadores de grandes capacidades, el BSC es parte de la red nacional de supercomputación. Y esta red ofrece a los proyecctos científicos no sólo tiempo de cálculo, sino tiempo de almacenamiento de grandes cantidades de datos.

Cuando se termine de inaugurar el Mare Nostrum 5 será un momento importante para España y para Europa, porque significa que «la UE se pone las pilas», colocándose -en supercomputación e Inteligencia Artificial- al nivel de grandes potencias como EEUU o China que estan por delante.
¿Que investigaciones lleva a cabo el BCS? Muchos cálculos de ingenieria, también muchos estudios relacionados con el clima y el calentamiento global, y otros muy destacados son los de biomedicina, genómica, a los que se dedica mi departamento.
Sus investigaciones se centran en comprender los organismos vivos mediante métodos teóricos y de computación. Los superordenadores han sido capaces de hacer una simulación de la evolución del Universo observable, con más de un billón de galaxias. Pero teniendo en cuenta que cada cuerpo humano tiene aproximadamente 30 billones de células, que cada célula alrededor de 42 millones de proteínas, que el genoma humano tiene unos 20.000 genes codificantes, las interacciones entre ADN, ARN y proteínas, los nuevos descubrimientos en epigenética… ¿no es mucho más complicada, mucho más multifactorial, la tarea de la supercomputación de los sistemas vivos?
(Risas) Sí, mucho más. Yo esto lo comento medio en serio, medio en broma con mis compañeros que usan la supercomputación para problemas de cosmología o de física de altas energías, y que hacen investigaciones espectaculares y con aparatos muy sofisticados. Pero en el fondo, ellos están lidiando con una cantidad de datos más limitada, y además son datos muy homogéneos y con un margen de error muy controlado, y sobre una realidad que es más sencilla, con menos factores actuando.
Como bien dices, un sistema biológico es mucho más complejo, el número de factores actuando -células, genes, tejidos, estados fisiológicos…- es mucho mayor, y para colmo, los datos que nosotros manejamos son mucho más inhomogéneos, y sometidos a mucho más nivel de error. Por ejemplo las muestras tomadas de pacientes en los hospitales, están sometidas al error del clínico que toma la muestra, al del técnico de laboratorio que hace los ensayos, faltan datos… Nuestros datos son más complejos y también de menor calidad. Y por eso, la supercomputación en biomedicina es mucho más complicada que para otras disciplinas como la física. La frontera del conocimiento son los problemas en biología.
La supercomputación en biología es más compleja
Buena parte de sus investigaciones se centran en generar gemelos digitales, modelos computacionales que sean capaces de replicar de la forma más exacta posible el complejo funcionamiento de los sistemas vivos. Por ejemplo, de un tumor. ¿Es así? ¿En qué consisten estos gemelos digitales? ¿Es posible lograr una simulación hiperrealista de un cuerpo humano, de cada persona, con su configuración genética concreta, para predecir y ajustar tratamientos oncológicos personalizados?

Antes que nada, aclarar que el término de los gemelos digitales es común en ingeniería. En la industria, es usual crear modelos de simulación de un coche o una máquina que imita su funcionamiento, para ver cómo se comporta en determinadas situaciones, cuándo se desgasta o se rompe una pieza, para predecir qué va a pasar si hago un cambio u otro.
En el BSC tratamos de generar gemelos digitales del clima, de ciudades, de epidemias. Evidentemente, aún no tenemos un gemelo digital de un cuerpo humano completo que nos sirva para emular cómo se comportará un individuo en una intervención quirúrgica o algo por el estilo. Lo que hacemos en biocomputación es funcionar por aproximaciones. Hacer aproximaciones por capas que emulen lo mejor posible las diferentes capas de complejidad.
Por ejemplo, sabemos que podemos hacer simulaciones de manera muy realista de procesos atómicos y moleculares: una proteína uniéndose a un fármaco o al ADN. Este tipo de simulaciones se usan en la industria farmacéutica continuamente.
Subiendo un nivel, comenzamos a tener simulaciones de órganos bastante realistas Por ejemplo del corazón: con sus cavidades, sus flujos, sus contracciones, su electrodinámica. Estos modelos se usan en cirugía para planear las operaciones de arritmias. Se estudia y se toman datos del corazón real, se hace un modelo computacional, y se planea la intervención sobre ese modelo personalizado. No los llamaría hiperrealistas, pero sí lo suficientemente realistas como para dar sugerencias sobre cómo intervenir.
Y nosotros también hacemos modelos a nivel celular, por ejemplo simular un tumor, algo que es supercomplicado por la cantidad de variantes que intervienen: el tipo de células malignas, cómo se distribuyen y van progresando, el tipo de células del tejido y la intervención del sistema inmune, si hay metástasis o no… Pero usamos esas simulaciones para anticipar la respuesta del tumor a fármacos. A estas simulaciones les queda aún mucho por desarrollar, pero ya empiezan a tener una capacidad predictiva.
La biocomputación no sólo vale para desarrollar terapias o tratamientos, sino para el conocimiento. Por ejemplo, es posible cultivar en el laboratorio algunos órganos, llamados organoides (una especie de miniórganos que tienen funcionalidad) a partir de células madre con capacidad de diferenciarse. Y esos modelos se pueden usar para ver cómo afecta una mutación o un fármaco. Y con ordenador podemos hacer una simulación en paralelo de estos organoides para expandir su capacidad. No sólo para predecir, sino también para estudiar y comprender por qué un fármaco o una mutación tienen el efecto que tienen, qué rutas moleculares usa una biomolécula para generar su efecto.
Nos movemos en estas aproximaciones: atómicos, celulares y de órganos, pero no tenemos un «gemelo digital» integral de cada cuerpo humano. Eso es lo que nos gustaría acabar teniendo, pero aún no está al alcance. Ya veremos cuántos años nos cuesta: hay quien dice que ese horizonte está a veinte años y hay quien dice cincuenta.
En biocomputación funcionamos por aproximaciones
Otra aplicación de la supercomputación a la biomedicina hace referencia al desarrollo de vacunas. Si conocemos la configuración tridimensional de las proteínas de un patógeno, por ejemplo de la espícula del coronavirus o los antígenos del virus del SIDA… ¿podemos «diseñar» en el superordenador los anticuerpos que mejor se van a unir a ellos de manera específica, y luego diseñar los genes que van a producir esos anticuerpos, para insertarlos en una vacuna?
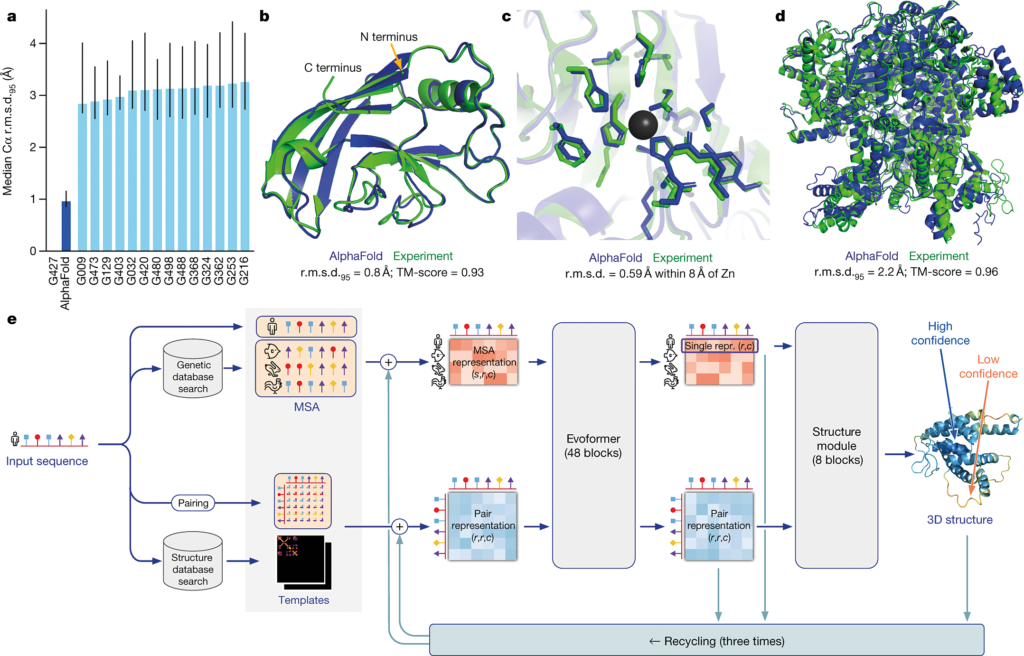
Si me hubieras hecho esta pregunta hace tres años, te hubiera dicho «esto es casi imposible, nos queda mucho». Pero después de que Jumper y Hassabis desarrollasen el sistema de inteligencia artificial AlphaFold -que es capaz de predecir estructuras de proteínas a partir de su secuencia de aminoácidos, y que ha logrado una altísima precisión- esto ya está al alcance. De hecho se están mejorando este tipo de computaciones, y una vez conocidas las claves, hay laboratorios públicos que ya pueden predecir la estructura de las proteínas a partir de su secuencia de un modo casi equivalente a su resolución estructural.
El siguiente problema es la interacción entre proteínas. Estos software funcionan muy bien para interacciones estables, como las que se dan entre una proteína y un ribosoma. Interacciones entre superficies muy pequeñas, como las que se dan entre antígeno y anticuerpo son más difíciles de predecir, pero se han hecho enormes progresos en los dos últimos años.
Esto resuelve una parte de lo que me has preguntado y nos sirve para diseñar anticuerpos, pero esta es la parte menos útil, porque es la respuesta humoral del sistema inmune que es la menos potente. Lo que nos interesaría más es incidir en modular la respuesta celular, cómo elegir los fragmentos de un virus o un patógeno para que desencadenen una respuesta inmune celular potente, uniéndose al receptor HLA de las células T. Pero eso ya es una interacción entre tres cuerpos (antígeno-anticuerpo-HLA de las células T) y ese es un problema computacional muy complejo. Entonces lo que hacemos es generar muchos modelos, generar y probar hipótesis, y eso hace más fácil hacer experimentos.
Esto lo hacemos constantemente para diversos equipos de investigación y para muchas compañías: generar hipótesis sobre qué fragmentos de la proteína de un patógeno van a desencadenar una mejor respuesta inmune. Te pongo un ejemplo: las terapias inmunológicas contra el cáncer están siendo toda una revolución en oncología. Pues el ideal sería predecir por biocomputación, para cada persona, con este tipo de mutaciones concretas en su tumor, qué produce estas proteínas mutadas en la superficie de sus células malignas (marcadores), y voy a diseñar los mejores anticuerpos, la respuesta humoral más adecuada. De todas las mutaciones que tiene este cáncer -porque suelen tener varias simultáneamente- voy a seleccionar las más adecuadas para desencadenar la respuesta inmune más eficaz, que no vaya contra las células sanas, sino que vaya dirigida sólo a las mutaciones seleccionadas y por tanto sean capaces de detener el cáncer.
Para esto primero hay que hacer un análisis del genoma, luego ver qué proteínas marcadoras van a producir, luego predecir qué regiones de esas proteínas van a tener un mayor potencial inmunogénico uniéndose al HLA. Y esto es algo que ya estamos haciendo en colaboración con el hospital Vall d’Hebrón.
Su colega del BCS Rachel Lowe trabaja en gemelos digitales que ayuden a predecir epidemias, incluso de patógenos emergentes con capacidad pandémica. Con modelos computacionales que tienen en cuenta las características del agente patógeno, el acervo genético de la población, los factores medioambientales… e incluso las buenas o malas decisiones políticas de los gobiernos en cuanto a la salud pública. ¿Es así? ¿Se puede simular a este nivel de detalle la evolución epidemiológica de una enfermedad?

Desde luego, y es fascinante. Hemos hecho progresos enormes. Son modelos computacionales que son capaces de ligar los datos del clima, las variaciones estacionales, el impacto del cambio climático, y cómo afectan a cómo se distribuyen determinados vectores -por ejemplo los mosquitos-, en qué proporciones estos mosquitos son portadores del patógeno, cómo se distribuyen en distintas áreas de una ciudad o una región atendiendo a la densidad de su población, cómo diferentes grupos sociales son más o menos susceptibles de enfermar, y cómo diferentes decisiones políticas sobre salubridad e higiene, restricciones a la movilidad, o distribución de vacunas influyen positiva o negativamente en la evolución epidemiológica de la epidemia.
Y estos modelos de Rachel no son sólo importantes para países en desarrollo, sino para nuestro entorno, porque el cambio climático ya está haciendo que haya enfermedades emergentes en Europa o en España. Donde antes no llegaban vectores animales que portaban determinados patógenos tropicales (por ejemplo la fiebre del Nilo), la subida de las temperaturas hace que los haya en estas latitudes. La multiplicación del transporte o del turismo global hace que puedan llegar.
Aplicar el ChatGPT para biomedicina
¿Qué otros desarrollos permite la supercomputación en el terreno de la biomedicina? ¿Qué perspectivas abre esta disciplina para la humanidad?

Yo siempre digo que la supercomputación no va a resolver por sí sola ninguna enfermedad, pero que difícilmente los problemas de la medicina, del cáncer o de la salud pública se van a resolver sin la supercomputación. Porque se trata de problemas con tantos datos, tantas variantes, que necesitan de estas herramientas para hallar soluciones.
Las cosas más emocionantes que están pasando en este área es la revolución en los modelos del lenguaje, como el famoso ChatGPT. Además de que es muy entretenido para otras tareas, a nosotros nos interesa su aplicación a la biomedicina, porque nos ofrece una vía para descifrar y procesar la enorme cantidad de información contenida en los Medical Records, los historiales clínicos. Estos historiales los elaboran los médicos para su uso profesional, pero si somos capaces de digerir ese «big data», esa enorme cantidad de información acumulada, podríamos tener el mayor experimento clínico que ha hecho la humanidad. Estamos hablando de una inmensa cantidad de datos sobre miles de millones de pacientes en todo el mundo.
Son lo que llamamos los ‘Large Language Models‘ (LLM), algoritmos de lenguaje colosal, o modelos de lenguaje de gran tamaño, que permiten procesar grandes cantidades de información -a menudo de textos no estandarizados, como un historial clínico- y transformarlos en datos que pueden ser procesados por supercomputación
Esta misma semana haremos público un corpus, que se llama CARMEN, de textos médicos anotados, que es el mayor recurso mundial disponible para entrenar a las supercomputadoras en descifrar estos Medical Records.
Y esta aplicación de los Large Language Models lo podemos hacer para muchos otros campos. El ChatGPT genera un nuevo texto sintético, y eso mismo lo podemos aplicar para generar proteínas completamente nuevas. Entrenar a los superordenadores a que a partir de un alfabeto -20 aminoácidos- produzca un texto coherente -una secuencia de proteínas- que se pliegue correctamente y genere una estructura funcional que tenga funciones novedosas, fuera de las proteínas conocidas en la naturaleza. Podemos expandir el universo de proteínas a nuestro alcance enormemente, generando biomoléculas de propiedades innovadoras. Y esto abre un abanico de posibilidades impresionante en genómica y en biomedicina.
Carlos dice:
Oyes, el maestro Alfonso Valencia me ha abierto nuevos horizontes para desarrollar productos gratuitos para sourceforge https://sourceforge.net/ en beneficio de toda la humanidad
Gracias maestro