
Dos estudios independientes abordan mediante la edición genética una terapia para curar la rara enfermedad del envejecimiento acelerado.
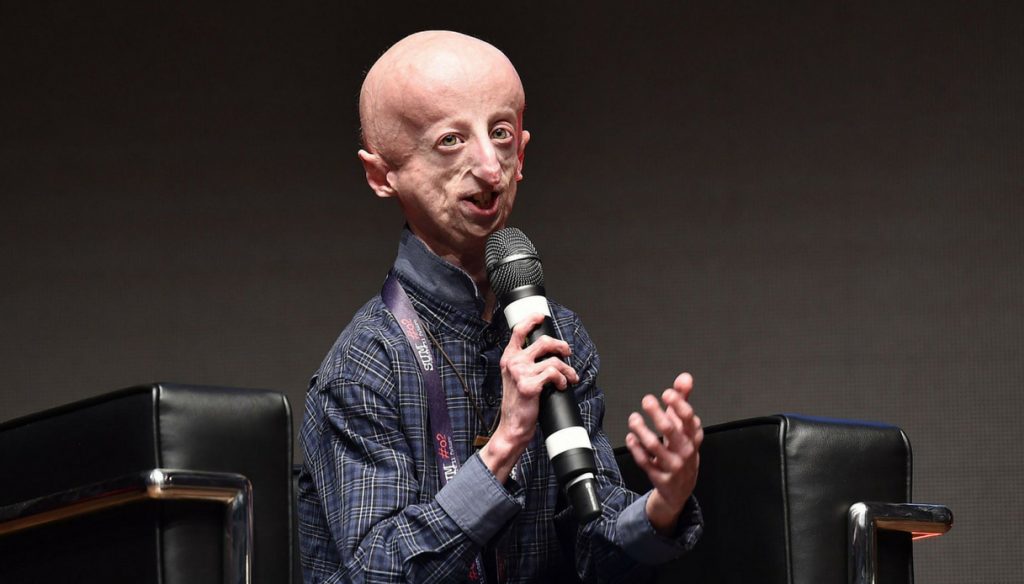
Cuando Sammy Basso nació en 1995 en Schio, una pequeña ciudad del norte de Italia, nada parecía indicar que en lo más profundo de sus genes yacía enterrada una maldición: el extremadamente raro transtorno genético de la progeria o síndrome de Hutchinson-Gilford. Es la enfermedad de los «niños ancianos»: quienes la padecen envejecen a cámara rápida y su esperanza de vida no suele superar los 13-14 años. A sus 23 años, Basso es uno de los casos más longevos del mundo.
Hay quien piensa que algo tiene que ver su longevidad con la esperanza, con la tenacidad y la perseverancia. Sammy no solo se resiste a la enfermedad. Lleva toda su vida luchando -literalmente- por comprenderla, curarla y erradicarla. Siendo niño, conoció a uno de los principales expertos del mundo en envejecimiento, el español Carlos López Otín, y decidió que él también quería ser biólogo molecular. Ahora, tras licenciarse en la Universidad de Padua, ha realizado la parte experimental de su trabajo fin de carrera -centrado en la investigación de esta dramática enfermedad- en la Universidad de Oviedo junto a su mentor.
Sammy Basso, ahora ya asturiano de adopción, forma parte de uno de los dos equipos del mundo que -de forma simultánea y con enfoques muy similares- proponen un tratamiento de la progeria que podría ser el primero con efectos permanentes, al actuar directamente sobre el gen mutado mediante técnicas de edición genética. Ambos grupos -uno liderado por el catedrático Carlos López-Otín de la Universidad de Oviedo, y otro encabezado por el mundialmente famoso Juan Carlos Izpisúa-Belmonte del Instituto Salk de California- han publicado en la revista Nature Medicine dos estudios independientes que se basan en la conocida herramienta de edición genética CRISPR/Cas9, cuyo mecanismo molecular fue descubierto originalmente por el microbiólogo alicantino Francis Mojica.
La progeria de Hutchinson-Gilford está causada por una mutación en el gen denominado LMNA, que provoca la acumulación de una proteína tóxica en el núcleo de las células. Recientemente se habían desarrollado terapias exitosas con resultados positivos para frenar este síndrome, pero el tratamiento propuesto en ambos trabajos es totalmente diferente. No buscan paliar o retrasar la progeria. Buscan editar y corregir el ADN de las células -mediante el sistema CRISPR, un «editor de texto» genético- para borrar la enfermedad de los cromosomas. Persiguen la curación completa de los pacientes.
«El objetivo es modificar el gen LMNA para evitar así la producción de la proteína tóxica», afirma Izpisúa-Belmonte. Los autores de ambos trabajos han ensayado este sistema de edición génica en un modelo murino (ratones modificados genéticamente para que expresen la enfermedad) generado previamente en el laboratorio del equipo asturiano de López-Otín para estudiar esta patología humana. Como explica Olaya Santiago-Fernández, coautora del estudio de la Universidad de Oviedo, «el uso de esta técnica en ratones con progeria da lugar a un aumento en su esperanza de vida y a una mejoría de los síntomas de envejecimiento acelerado».
Pero la dificultad de esta misión es enorme. ¿Cómo corregir el gen LMNA erróneo responsable del envejecimiento en los genomas de los aproximadamente 30 billones de células del cuerpo humano?. Los investigadores han introducido en virus modificados las herramientas CRISPR, con secuencias diseñadas para reconocer específicamente el gen LMNA y modificarlo para evitar la producción de la proteína tóxica. En los ratones de los ensayos, el grado de eficacia ha sido variable pero esperanzador. En el hígado, el órgano con mayor éxito, la estrategia ha servido para modificar el 14% de todas sus células, lo cual ya ha servido para aumentar un 25% la longevidad de los ratones afectados por el síndrome.
«Esto demuestra que no es necesario modificar el 100% de las células del organismo para corregir la enfermedad», afirma Olaya Santiago. Este enfoque demuestra por primera vez la capacidad del sistema CRISPR/Cas9 para afrontar enfermedades sistémicas, que afectan no a un tejido o a un órgano concreto, sino a todo el organismo. «Las dos investigaciones son fantásticas» y suponen la primera demostración de que el CRISPR puede funcionar de manera multiorgánica, dice Marc Güell, químico en la Universidad Pompeu Fabra de Barcelona.
Y desde luego, estos dos trabajos son un gran paso adelante en la posible aplicación de las terapias de edición génica con CRISPR/Cas9 para el tratamiento de enfermedades genéticas y congénitas, hasta ahora incurables como es el caso de la progeria de Hutchinson-Gilford.
La comunidad científica biomédica está entusiasmada y ha celebrado la enorme valía de estos dos trabajos, pero tanto sus autores como sus colegas se atienen a la cautela y a la moderación.
“Dado que la progeria es una enfermedad devastadora y que no hay otras terapias disponibles, no creemos que se necesite mucho tiempo para comenzar ensayos clínicos en humanos. Como con cualquier técnica, siempre habrá algunas preocupaciones sobre su seguridad, pero en este caso el resultado positivo es mucho mayor que los pequeños efectos negativos”, declara el bioquímico Juan Carlos Izpisúa, del Instituto Salk de California, a El País.
Otros, como el pionero de la edición génica en España, Lluis Montoliu, recuerdan que es necesario afinar los sistemas CRISPR para humanos, para garantizar la eficacia y la seguridad necesarias para proceder a ensayos sistémicos.
La cura de la progeria mediante edición genética aún no es una realidad, pero se siente al alcance de la mano. Sammy Basso sonríe. No sabe si llegará a tiempo para él, pero es feliz de saber que es parte de la vanguardia científica que está abriendo el camino de erradicar esta enfermedad. “Hace unos años, incluso hace unos días, nadie podría haberlo imaginado, pero ahora sí. Ahora podemos tener esperanza en llegar a una cura. No sé si me ayudará a mí, pero por primera vez puedo creer que, en el futuro, los niños con progeria podrán vivir una vida normal”. Ninguna cautela puede borrar la esperanza de su joven corazón.